KROMOZOM
Kromozom, (Yunanca'dan: chromos = renk + soma = vücut)
Her canlı gibi insan da trilyonlarca hücreden meydana gelir. Hücre, bitkisel ya da hayvansal her türlü yaşam biçiminin en küçük birimidir. Her hücre bir sitoplazma ve çekirdekten meydana gelir. Çekirdeğin içinde ise kromozom adı verilen ipliksi parçalar bulunur. İnterfaz evresinde kromatin ağı şeklinde bulunan DNA, mitoz bölünmenin profaz evresinde kısalıp kalınlaşmaya başlar ve metafaz evresinde en kısa duruma gelir. yaklaşık 10.000 kat kısalmış haliyle ışık mikroskobunda 100 lük objektifte incelenebilir. Kromozomlar, İ, V, J harfleri gibi biçimlerde görünür ve boyutları mikronla ölçülür. Kromozomların sayısı canlı türleride değişiklik gösterir. Örneğin sirke sineğinde 8, kurbağada 26, farede 42, köpekte 78 kromozom vardır. İnsanın kromozom sayısı ise 46'dır. 22'si çift otozom kromozomdur. İnsan hücresinde 1 çift de eşeysel kromozom bulunur ve toplam sayı 46 eder. Eşey kromozomları kadınlarda XX, erkeklerde XY dir. Kromozomlar, molekül yapıları çok iyi bilinen DNA (deoksiribonükleik asit) zinciri ile histon denilen protein zincirinden oluşur. DNA zincirleri de özgül proteinleri sentezlemekle görevli gen adı verilen birimlerden oluşur.lfgmfjnkjaldfjgjfgllsoeojgmldşdişlf,p
Döllenme sırasında annenin yumurtasındaki 23 kromozom, babanın spermindeki 23 kromozomla birleşir. İşte bu 46 kromozom insanın yaşamında belirleyici rol oynar. Kromozomlarda yer alan ve sayıları 25 bin ile 30 bin arasında olduğu tahmin edilen genlerin oluşturduğu zincir, kişinin göz renginden boyuna, yaşam süresinden yakalanacağı hastalıklara kadar pek çok şeyi programlar. Bu genetik programlar, nükleotid denen (A, T, C, G) yapıların farklı dizilimleriyle şifrelenir. . Kromozomların mikroskop altında incelendiği bilim dalına sitogenetik adı verilir. Bu şekilde kromozom sayısında (Ör. Down Sendromunda 47, Turner sendromunda 45) veya yapısındaki değişiklikler(Parça eksilmesi -delesyon veya iki kromozom arasında parça değişimi translokasyon gibi)bu şekilde saptanabilir. Ancak kromozomlardaki bir değişikliğin mikroskopta görülebilmesi için en az 3milyon nükleotidlik bir kısmın değişmesi gerekir, daha küçük değişiklikler ancak moleküler genetik yöntemlerle incelenebilir.
Her insan hücresinde yaşamın yapı taşları kabul edilen 23 çift kromozom bulunuyor. Gen bilgilerini taşıyan ip biçimindeki kromozomlar uç uca eklenseydi 1,8 metrelik bir kordon oluştururdu. Kromozomların bozuk oluşumu sonucu, insanın yaşamında değişik dönemlerde, çeşitli hastalıklar ortaya çıkıyor. Bilim adamları, hangi kromozomun bozuk olduğunda hangi hastalığa neden olduğunu biliyorlar.
Kromozom anomalileri; bir kromozomda meydana gelen yapısal ya da sayısal değişiklikleri gösterir. Genellikle mayoz ve mitozu izleyen hücre bölünmesi sırasında meydana gelen hatalardan kaynaklanırlar. Birkaç farklı türü bulunmakla beraber, genel olarak sayısal ve yapısal anomaliler olarak iki ana gruba ayrılırlar.
 Daha yüksek çözünürlüğe sahip sürüm bulunmamaktadır.
Chromosome.png
Daha yüksek çözünürlüğe sahip sürüm bulunmamaktadır.
Chromosome.png (200 × 287 piksel, dosya boyutu: 3 KB, MIME tipi: image/png)
| Description |
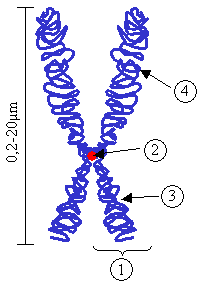
English: Chromosome. (1) Chromatid. One of the two identical parts of the chromosome after S phase. (2) Centromere. The point where the two chromatids touch, and where the microtubules attach. (3) Short arm (4) Long arm.
Deutsch: Abb. 1. Chromosom: 1. Chromatid, 2. Centromer (auch Spindelfaseransatzstelle genannt), der Punkt, an dem sich die beiden Chromatiden berühren, 3. Kurzer Arm. (p-Arm), 4. Langer Arm. (q-Arm)
日本語: 染色体: (1) 染色分体:染色体に含まれる2つの同一の部分のうちの片方 (2) セントロメア: 2つの染色分体が接合する場所で、ここに微小管が結合する (3) 短腕 (4) 長腕
Nederlands: Afb. 1. Chromosoom: 1. Chromatide, 2. Centromeer, 3. Korte arm, 4. Lange arm
Please note: In Cytogenetics, chromosomes are always presented with the short arm on top, unlike in this image. An updated version is available (see below)
|
| Source |
Nupedia, then en.wikipedia
|
| Date |
Uploaded on en.wikipedia 21:18, October 4, 2002
|
| Author |
Magnus Manske
|
| |
|
| Other versions |
This updated version is "upright": In Cytogenetics, chromosomes are always displayed with the short arm on top. 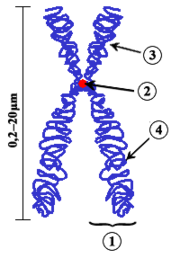 |
Sayısal anomaliler
-
Çift kromozomlardan birinin kaybolması (monozomi) ya da bir çiftte ek olarak bir tane daha kromozomun bulunması (trizomi) olarak tanımlanır. Sayısal anomalilere en bilinen örnek, "Trizomi 21" (üç tane 21. kromozom) olarak da bilinen "Down sendromu"dur. Monozomi olarak ise, eşey kromozomlarından birinin olmaması durumu olan (45,X) "Turner sendromu" gösterilebilir.
Yapısal anomaliler
Kromozomun yapısal formunun değişmesiyle oluşan birkaç farklı türü vardır:
- İnversiyonlar: Kromozomdaki bir kısmın kopup, ters dönüp daha sonra tekrar aynı yere birleşmesiyle meydana gelen düzensizliklerdir.
- Ring kromozomlar: Kromozomun bir parçasının kopup, daha sonra halka şeklinde kendiyle birleşmesiyle meydana gelen düzensizliklerdir. Genetik materyal kaybı olmadan ya da olarak gerçekleşebilir.
Kalıtımı
Kromozom anomalileri daha çok, kalıtılmayan fakat, yumurta ya da spermde meydana gelen hatalardan kaynaklanırlar. Vücudun bütün hücrelerinde görülebileceği gibi, bazen bazı hücrelerin normal bazılarının anomalili olmasıyla (mozaizm) da karşılaşılabilir. Kromozom anomalileri, aileden gelebileceği gibi, aniden "de novo" olarak da gerçekleşebilir. Bu, ailesi normal olan çocuğun neden anomalili olabileceğini açıklar.
Ploitlik (İng., ploidy), bir hücredeki kromozom sayısının, o hücredeki farklı kromozomlardan birer tanesi ile oluşturulan temel setteki kromozom sayısı ile bir rasyonel sayının çarpımına eşit olması durumudur [1]. Diğer bir deyişle ploitlik, temel kromozom takımının (ve sayısının) ilgili hücredeki tekrarlama derecesidir [2].
İlgili terimler Genetikte, bir temel kromozom takımı (ve sayısı) n ile gösterilir. Ploitliği de n ile çarpıldığında hücredeki toplam kromozom sayısını veren rasyonel sayının bir tam sayı olup olmamasına göre sınıflandırmak ve çeşitlendirmek mümkündür:
-
- Öploitlik (İng., euploidy) : n'in tam katları kadar kromozom içerme durumu.
-
- Öploit kromozom sayısı → öploit hücre → öploit canlı
- Haploitlik (İng., haploidy) : n kadar kromozom içerme durumu; monoploitlik (İng., monoploidy).
-
- Haploit kromozom sayısı → haploit hücre → haploit canlı
- Diploitlik (İng., diploidy) : 2n (2 x n) kadar kromozom içerme durumu.
-
- Diploit kromozom sayısı → diploit hücre → diploit canlı
- Poliploitlik (İng., poliploidy) : 2'den fazla n kadar kromozom içerme durumu.
-
- Poliploit kromozom sayısı → poliploit hücre → poliploit canlı
- Triploit (3n), tetraploit (4n), pentaploit (5n), hekzaploit (6n), heptaploit (7n), oktoploit (8n), nonaploit (9n), dekaploit (10n).
- Anöploitlik (İng., aneuploidy) : n'in tam katları olmayan kadar kromozom içerme durumu; benzer terim, heteroploitlik (İng., heteroploidy).
-
- Anöploit kromozom sayısı → anöploit hücre → anöploit canlı
- Hipoploitlik (İng., hypoploidy) : n'in tam katından biraz daha az sayıda kromozom içerme durumu.
-
- Hipoploit kromozom sayısı → hipoploit hücre → hipopploit canlı
- Hiperploitlik (İng., hyperploidy) : n'in tam katından biraz daha fazla sayıda kromozom içerme durumu.
-
- Hiperploit kromozom sayısı → hiperploit hücre → hiperploit canlı
Poliploitlik, bir hücrenin ya da canlının kendisindeki her bir kromozomun (diğer bir deyişle de temel kromozom takımı n'in) ikiden fazla kopyasına sahip olması durumudur. Canlıların çoğunluğu diploit olsa da hücre bölünmesinin olması gerektiği gibi gerçekleşmemesi sonucu, poliploit hücre ve canlılar ortaya çıkabilir.
Poliploitlik ve yeni türler
Poliploitlik yeni türlerin ortaya çıkmasına neden olabilir. Bunun en ünlü örneklerinden birisi Spartina cinsi bitkide yaşanmıştır:
-
- 19. yüzyıla kadar sadece Spartina maritima (2n [AA] = 60 kromozom) adlı Spartina türünün yaşadığı İngiltere'de, 1829'dan itibaren Kuzey Amerika kökenli ve farklı bir Spartina türü olan Spartina alterniflora (2n [BB] = 62 kromozom) görülmeye başlanmıştır.
- Spartina maritima ile büyük olasılıkla gemi yoluyla İngiltere'ye gelmiş olan Spartina alterniflora arasındaki döllenme sonucunda, kendisi üreyemeyen bir melez olan Spartina x townsendii ortaya çıkmıştır.
- Daha sonraları, 1892'de, bölgede farklı bir Spartina türü keşfedilmiş, bunun yeni bir tür olduğu anlaşılmış ve Spartina anglica diye adlandırılmıştır. Araştırmalar, bu türün Spartina x townsendii melezinde oluşan poliploitlik sonucu ortaya çıktığını göstermiştir: Spartina x townsendii 61 kromozama sahipken (n+n [A+B]) ve üreyemezken, Spartina anglica 122 kromozoma sahip, tetraploit (4n [AABB]) ve üreyebilen bir türdür.
Poliploitliğin dikkat çektiği bir başka bitki cinsi de çilek, yani Fragaria'dır:
-
- Farklı 7 kromozomdan oluşan temel bir set tüm çilek türlerinde ortak olarak bulunurken, farklı türler farklı poliploitlik derecesi gösterirler.
- Bu şekilde, yirmiden fazla çilek türünü diploit, tetraploit, hekzaploit, oktoploit ve dekaploit oluşlarına göre 5 grup içinde sınıflandırmak mümkündür.
- İstisnaları olsa da kabaca, daha çok kromozom içeren çilek türlerinin daha dayanıklı olduğundan ve hem bitki, hem de meyve olarak daha büyük geliştiğinden bahsedilebilir.
Hayvanlar âlemine ait bir poliploitlik örneği ise Afrika tırnaklı kurbağasıdır:
-
- Bilimsel adı Xenopus laevis olan bu kurbağa, Xenopus cinsinin diğer çoğu üyesi gibi, tetraploittir.tr:Ploitlik
|









